
服务内容
第一性原理计算
通过原子与电子尺度模拟,开展材料性能预测、结构稳定性分析、机理研究与方案优化,为新材料设计与技术开发提供可靠的理论依据
服务简介
可提供晶体、分子、表面、缺陷、掺杂及界面体系的第一性原理计算,服务内容包括结构优化、电子结构分析、吸附与反应机理研究、磁性分析、材料性质计算以及热力学与动力学稳定性评估。
针对不同研究目标,可灵活设计计算方案,满足论文研究、项目申报、课题支撑及企业研发等多类需求。
针对不同研究目标,可灵活设计计算方案,满足论文研究、项目申报、课题支撑及企业研发等多类需求。
可开展内容
支持金属、半导体、二维材料、催化材料、电池材料、储能材料及复合体系等多种模型。
可围绕材料筛选、性能预测、掺杂调控、界面作用、表面吸附、扩散行为及反应路径等方向开展定制化研究。
同时支持从模型搭建、参数设置、计算执行到结果整理与图表输出的一站式服务。
可围绕材料筛选、性能预测、掺杂调控、界面作用、表面吸附、扩散行为及反应路径等方向开展定制化研究。
同时支持从模型搭建、参数设置、计算执行到结果整理与图表输出的一站式服务。
输出成果
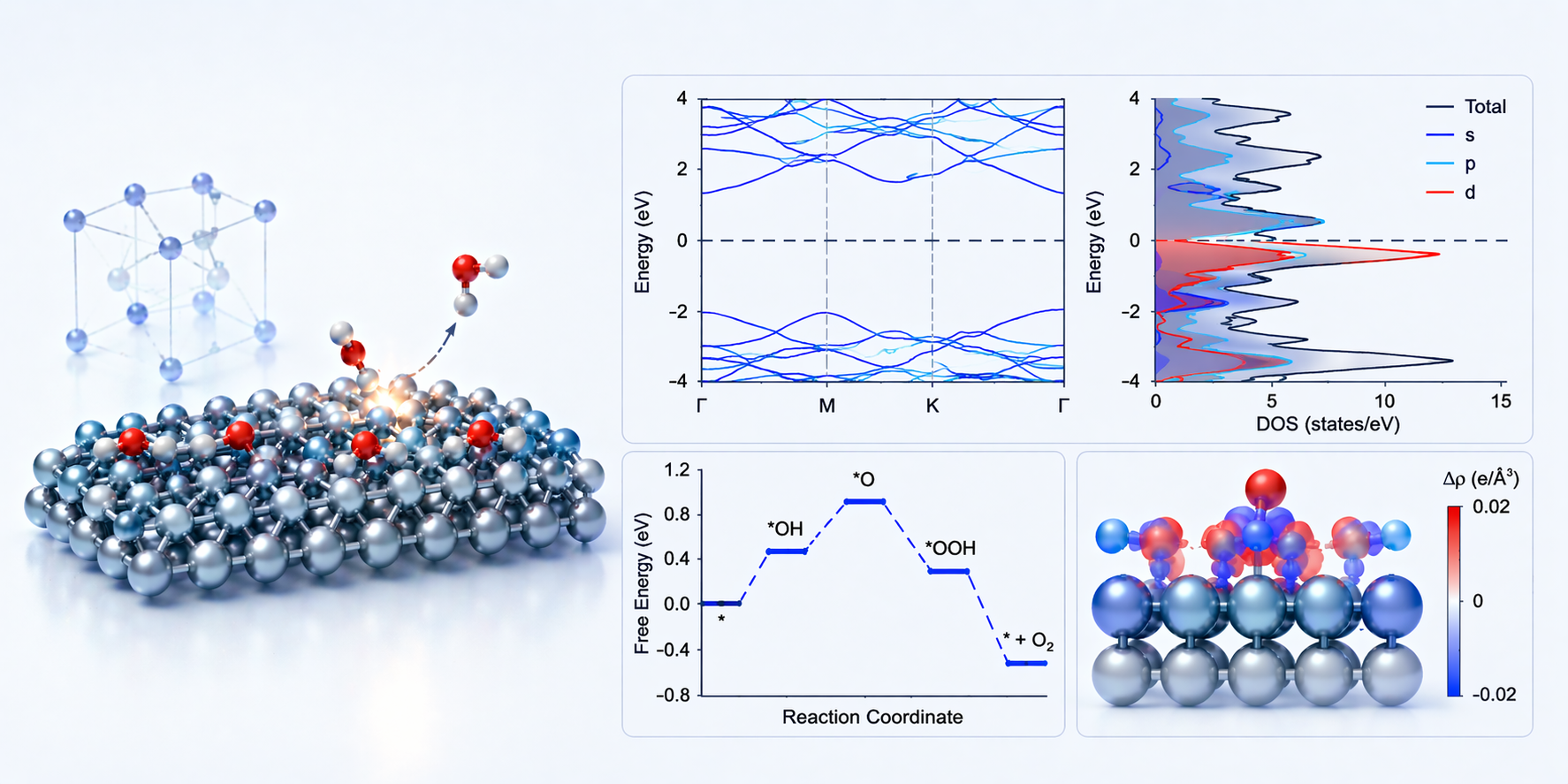
可提供结构参数、总能、形成能、吸附能、能带、态密度、电荷分析、磁矩、反应能垒等关键结果,并配套输出结构图、能带图、DOS图、电荷差分图、反应路径图等常用图件。
根据需求,还可提供分析报告、原始输入输出文件及可复用绘图文件等服务,便于后续论文撰写、项目汇报和持续研究。
根据需求,还可提供分析报告、原始输入输出文件及可复用绘图文件等服务,便于后续论文撰写、项目汇报和持续研究。