
Battery
Latest Nature report on lithium-sulfur batteries! Quantum chemistry + machine learning design of lithium-sulfur precursors.
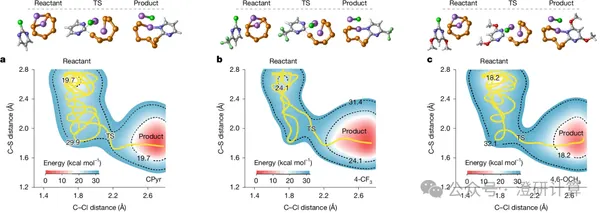
Molecular mediators in lithium-sulfur batteries are essentially a class of organic molecular additives added to the electrolyte. They interact with polysulfides to alter sulfur conversion pathways, reduce reaction polarization, and improve the Li₂S deposition process. Past molecular additive design focused more on functional groups, such as their ability to bind polysulfides or promote Li₂S formation. This Nature paper by Zhou Guangmin's team at Tsinghua University's Shenzhen International Graduate School focuses on the molecular skeleton of organic additives: even within the CPyr class of molecules, different substituents and substitution sites simultaneously affect the rate at which the molecule is activated by polysulfides, as well as the charge transfer capability of the activated mediator.
Read Original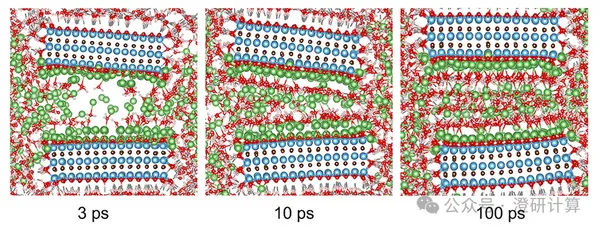
Other
AM: Water-mediated exfoliation achieves kilogram-scale MXene, DFT/MD analysis of Li⁺–H₂O hydrogen bond network
The challenge of large-scale MXene preparation lies in achieving both high yield and preserving large flake size and low defects. Traditional methods such as sonication, oscillation, or strong shearing can improve exfoliation efficiency, but they easily break up the layer fragments and introduce defects, thus affecting electrical conductivity, mechanical properties, and ion sieving. This paper in *Advanced Materials* by the teams of He Daping and Shen Jie from Wuhan University of Technology and Tan Rui from Swansea University proposes water-mediated exfoliation as an alternative to strong mechanical exfoliation. The article modulates the coordination environment of Li⁺ in the interlayer by water molecules, forming a Li⁺–H₂O hydrogen bond network, weakening interlayer electrostatic attraction and van der Waals interactions, thus achieving kilogram-scale, monolayer, large-size, low-defect MXene preparation. The theoretical calculations, combining DFT and MD, explain why water molecules promote interlayer expansion and why defect-free MXene channels exhibit higher Li⁺/Mg²⁺ selectivity.
Read Original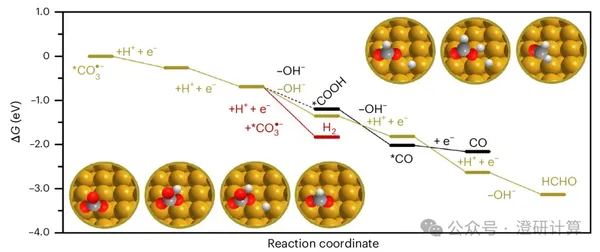
Catalytic
Nature Chemistry: The overlooked carbonate ion in CO₂RR actually restructures the interfacial water
CO₂ electroreduction commonly uses bicarbonate electrolytes. Past discussions of the reaction mechanism have typically focused on CO₂, metal cations, and the catalyst surface, with bicarbonate/carbonate ions often treated as buffer components or sources of carbon loss. This Nature Chemistry paper by Christopher S. Kley's team at the Helmholtz Centre for Materials and Energy in Berlin, Germany, reintroduces the discussion of carbonate ions within the context of interfacial reactions. Using in-situ ATR-SEIRAS, differential electrochemical mass spectrometry (DEMS), isotope labeling, and DFT calculations, the paper demonstrates that in Au-catalyzed CO₂RR, carbonate ions and their radicals can modulate the interfacial water structure, further influencing the competitive relationship between CO₂RR and HER.
Read Original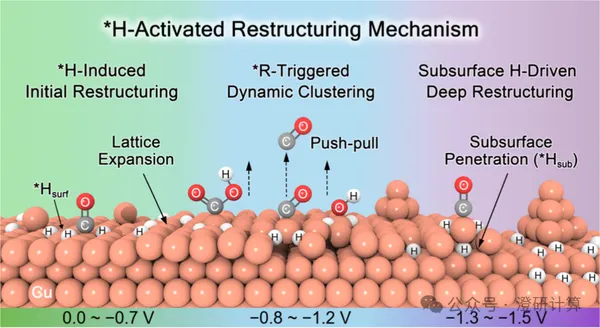
Catalytic
JACS: Why does the CO₂RR catalyst become increasingly coarse with each reaction? Adsorbed hydrogen provides a new explanation
In the electroreduction of CO₂, the Cu catalyst undergoes simultaneous reaction and reconstruction: the smooth surface gradually roughens, Cu atoms migrate, dissolve, and redeposit, ultimately forming low-coordination clusters. These new structures alter the active sites, directly affecting CO₂ activation, CO coupling, and the formation of multi-carbon products. Previously, Cu reconstruction was often attributed to CO adsorption or reduction potential, but this is insufficient to explain the continuous surface changes occurring over a wide potential range. A JACS paper by the team of Ling Chongyi, Wang Jinlan, and Li Qiang from Southeast University proposes that hydrogen adsorption is the key precursor: it first weakens the Cu–Cu bond and loosens the lattice, then allows intermediates such as CO and COOH to further trigger Cu atom migration and cluster formation.
Read Original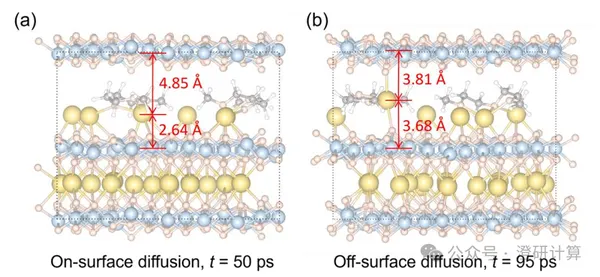
Other
Don't let your calculations go wrong because of your graphing! 8 top journal examples to teach you how to create advanced computational graphs
Theoretical calculations are an essential part of publishing papers, but producing good calculation results is only the first step. What impresses editors, reviewers, and readers is often the ability to organize complex physical images into a clear, beautiful, and logical diagram. In this issue, we've selected eight examples of computational graphs published in top journals such as Nature and Nature Energy, focusing on their strengths in graphic layout, color schemes, mechanism representation, and the integration of data and diagrams. We'll see how DFT, AIMD, MD, CI-NEB, COMSOL, or machine learning results can be transformed into more aesthetically pleasing, sophisticated, and publishable research graphs.
Read Original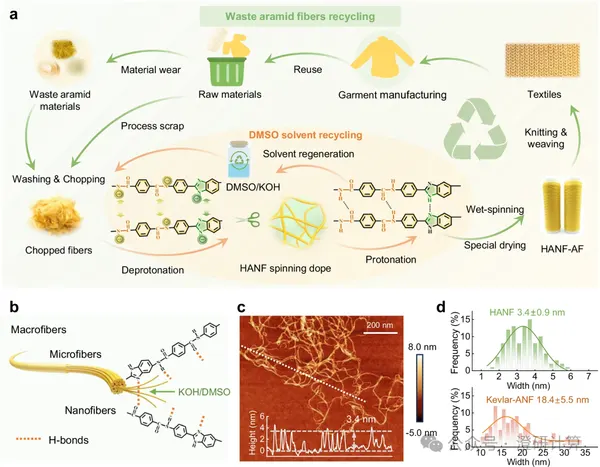
Other
Nature Communications: 3.4 nm aramid nanofibers woven into a thermal vest, H⁺-mediated assembly and theoretical calculation interpretation
Aerogel fibers are suitable for use in thermal insulation fabrics, but a long-standing trade-off between strength and thermal insulation has existed. Increasing porosity can reduce thermal conductivity; however, if the structure is too loose, the fibers cannot withstand stretching, bending, weaving, and sewing. This Nature Communications paper uses waste heterocyclic aramid fibers as raw materials to first prepare 3.4 nm aramid nanofibers, and then obtains knittable, high-strength, low-thermal-conductivity aerogel fibers through H⁺-mediated hierarchical assembly. The theoretical calculations focus on "why aramid can dissociate, how H⁺ induces assembly, and why hierarchical porosity simultaneously improves strength and thermal insulation," connecting molecular interactions, charge regulation, assembly processes, and macroscopic properties.
Read Original